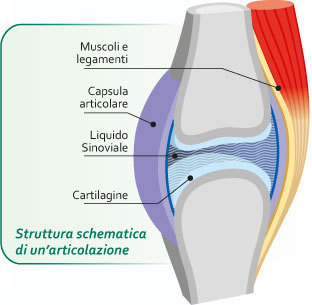
IL LES
Il LES è una malattia cronica infiammatoria che può colpire la pelle, le articolazioni, i reni, i polmoni, il sistema nervoso centrale e
periferico, il cuore e altri organi del corpo.
I sintomi più comuni sono l’arrossamento e l’infiammazione della pelle e l’artrite spesso accompagnata
da stanchezza e febbre.
Il decorso clinico del LES varia da forme molto leggere a forme gravissime e in genere la malattia alterna
periodi di benessere a ricadute. In alcuni casi la malattia può essere così grave da causare la morte, ma fortunatamente questo accade raramente. Inoltre le nuove terapie
a disposizione hanno migliorato la qualità della vita dei pazienti e ridotto grandemente il rischio di morte.
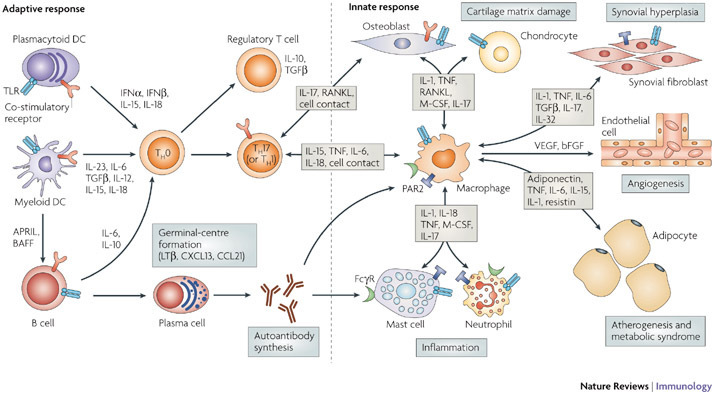
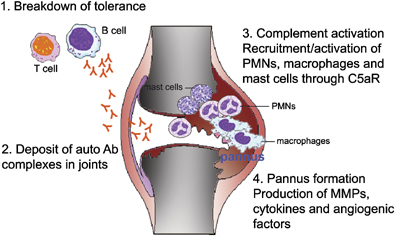
Il LES è una malattia autoimmune che si sviluppa quando il sistema immunitario, che normalmente ci protegge dai tumori e dalle infezioni,
attacca gli organi del corpo (si crea ciò che viene definito come la perdita della tolleranza immunologica). Questa perdita della tolleranza si manifesta con la
produzione da parte delle cellule del sistema immunitario di auto-anticorpi. Gli anticorpi sono proteine che attaccano i germi estranei all’organismo; si chiamano
auto-anticorpi quando attaccano le cellule dell’ organismo che li produce. Se l’attacco continua, altre cellule del sistema immunitario entrano in gioco. Questo porta alla
infiammazione dei vasi sanguigni (vasculite) e all’arrivo di cellule del sistema immunitario in diversi organi dove possono causare dei danni.
Non si sa perchè si sviluppi questa infiammazione ma è probabile che debbano essere contemporaneamente
presenti diversi fattori, in parte ereditari o famigliari e in parte ambientali (come ad esempio certi tipi di infezioni virali, la esposizione ai raggi ultravioletti del
sole, alle polveri di silicio e allergie a farmaci).
Recenti ricerche suggeriscono che i pazienti che si ammalano di LES possono avere un difetto nella
distruzione delle cellule vecchie o ammalate dell’organismo e che questo possa causare una anomala ed eccessiva stimolazione del sistema immunitario.
Deve essere ben chiaro che il LES non è una malattia infettiva e pertanto non è trasmissibile da
persona a persona. Non è una malattia ereditaria, trasmessa come tale dai genitori ai figli. Ciò che può essere trasmesso è la predisposizione a sviluppare la malattia.
MANIFESTAZIONI CLINICHE E DIAGNOSI
La diagnosi di LES può essere sospettata sulla base dei sintomi riferiti dal paziente ed è confermata da una serie di esami del sangue.
Il Lupus può dare manifestazioni a carico di qualsiasi organo o apparato.
Tipici sintomi del LES sono ad esempio:
- Febbre, stanchezza e perdita di peso
- Artrite in una o più articolazioni con durata anche di alcune settimane
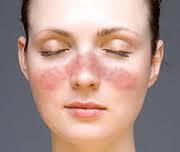
- Arrossamento della pelle del viso a forma di farfalla o altre lesioni della pelle
- Lesioni della pelle che compaiono immediatamente dopo una esposizione al sole
- Infiammazioni alla bocca o al naso che durano diverse settimane
- Perdita di capelli qualche volta a chiazze o al margine della attaccatura dei capelli
- Convulsioni, paralisi o disordini mentali
- Trombosi a diverse localizzazioni
- Aborti ripetuti in alcune pazienti
- Sangue o proteine nelle urine o alterazioni degli esami di funzionalità del rene
- Abbassamento delle cellule del sangue (anemia, riduzione delle cellule bianche o delle piastrine)
- Cattiva circolazione alle dita delle mani e dei piedi
Tutti questi sintomi possono svilupparsi nel corso di anni rendendo difficile la diagnosi del LES.
Tra le manifestazioni più frequenti e particolarmente caratteristiche vi sono le manifestazioni cutanee,
l’artrite, le sierositi (pleurite e pericardite), le alterazioni renali ed ematologiche. Le manifestazioni cutanee comprendono il tipico eritema a farfalla, il
lupus subacuto e quello cronico o discoide.
La fotosensibilità è molto frequente nei pazienti con LES e in molti pazienti l’esordio della malattia o una
sua riacutizzazione sono precedute dall’esposizione solare. La fotosensibilità può manifestarsi con gli eritemi tipici della malattia, con eritemi diffusi del
tutto aspecifici o con febbre, artralgie e altre manifestazioni. L’artrite è forse la manifestazione più frequente. Molti pazienti lamentano dolori accompagnati da gonfiore e
rigidità delle articolazioni. Le articolazioni più spesso interessate sono le piccole articolazioni delle mani, i polsi, le caviglie, le ginocchia, i piedi. Solo in una
piccola percentuale di pazienti si verificano deformità articolari. La pleura e il pericardio sono le membrane che avvolgono rispettivamente i polmoni ed il cuore.
L’infiammazione di tali membrane (pleurite e pericardite) determina dolore al torace e difficoltà a respirare.
A volte si hanno anticorpi diretti contro le cellule sanguigne; gli anticorpi contro i globuli rossi
determinano anemia, quelli contro i globuli bianchi e le piastrine determinano un calo di queste serie cellulari. Se i globuli bianchi sono bassi vi può essere una maggiore
suscettibilità alle infezioni. Se le piastrine sono molto basse si è più suscettibili ai sanguinamenti. L’infiammazione dei reni è riscontrabile nel 50% circa dei
pazienti con LES. Molto spesso si manifesta con alterazioni urinarie come proteinuria ed ematuria (che significa presenza di proteine e di globuli rossi nelle urine). Vi può
essere gonfiore alle caviglie, alle gambe, mal di testa, aumento della pressione, stanchezza. A volte è utile definire con esattezza il tipo di infiammazione renale e perciò
il medico proporrà al paziente una biopsia renale. Si tratta di un esame semplice che si esegue in anestesia locale e sotto guida ecografica, pungendo la parete toracica ed il
rene sottostante con un ago speciale che consente il prelievo di un piccolo frammento di tessuto renale. Questo viene poi analizzato al microscopio; è importante distinguere
una forma di glomerulonefrite dall’altra perché il trattamento non è sempre lo stesso e la prognosi è diversa.
L’interessamento neurologico, nelle sue forme più severe è raro; i quadri più caratteristici sono la
sindrome cerebrale organica e l’epilessia. La sindrome cerebrale organica si manifesta in maniera graduale o improvvisa con perdita di memoria e di altre funzioni
intellettive. E’ un quadro che può risolversi completamente se curato in modo corretto. L’epilessia è più frequente, può manifestarsi con forme di assenza o scosse
muscolari, accompagnate o meno da perdita di coscienza.
Il LES, soprattutto in fase precoce, può essere difficile da diagnosticare; infatti molte
manifestazioni sono estremamente aspecifiche. Ci sono alcuni esami del sangue che aiutano a fare la diagnosi.
Molto importanti sono gli anticorpi anti-nucleo (ANA) che sono presenti in tutti i pazienti affetti da LES.
Altri esami come la ricerca degli anticorpi anti DNA nativo (dsDNA) o degli anticorpi anti Sm sono più specifici e vengono usati per confermare la presenza del LES.
I livelli di certe proteine del sangue (ad esempio le proteine del complemento, che fanno parte
delle proteine del sistema immunitario) aiutano nella diagnosi e sono utilizzate per monitorare l’andamento della malattia.
Se sono presenti gli anticorpi anti- fosfolipidi, questo non solo aiuta nella diagnosi di LES,
ma indica anche il rischio di sviluppare certe specifiche complicanze della malattia. Queste includono un aumentato rischio di aborto e lo sviluppo di trombosi che
possono portare a infarti cerebrali o ad embolie polmonari.
TERAPIA
Il trattamento del LES dipende dalla sua gravità e dalla sintomatologia. Per dare al paziente la terapia più adatta è necessario che il medico
lo veda con regolarità, per riconoscere i sintomi iniziali delle possibili complicazioni della malattia.
In genere, per i sintomi che non sono pericolosi per la vita del paziente, come il dolore ai muscoli o
alle articolazioni, la stanchezza o le alterazioni della pelle, si usano farmaci sintomatici, come gli anti-infiammatori non steroidei. Molto utili per il loro
effetto immunomodulante, antiaggregante e per l’efficacia sulla sintomatologia cutanea ed articolare sono i farmaci anti-malarici (come l’idrossiclorochina o
Plaquenil).
Per le complicanze più gravi o che mettono a rischio la vita del paziente, come ad esempio per
l’infiammazione dei reni, il coinvolgimento del cuore o dei polmoni, le complicanze del sistema nervoso centrale o periferico, si utilizzano farmaci più aggressivi. In questi
casi si possono utilizzare alte dosi di cortisone (come il Deltacortene o il Medrol) e altri farmaci immunosoppressori come l’azatioprina, l’Endoxan (ciclofosfamide), il
Sandimmun Neoral (ciclosporina) o il CellCept (micofenolato).
Di più recente introduzione nel trattamento del LES sono i farmaci biologici come il Belimumab e il Rituximab che trovano impiego in casi selezionati(vedi sezione
farmaci biologici)
Spesso per controllare meglio la malattia o prevenire i danni agli organi i farmaci vengono
utilizzati in combinazione tra loro perché in questo modo sono più attivi del farmaco utilizzato da solo.
La terapia dipende dalla valutazione che viene fatta sul singolo malato pesando i possibili rischi e
benefici. Infatti molti farmaci immunosoppressori possono causare importanti effetti collaterali come ad esempio un aumentato rischio di infezioni, la nausea, il vomito, la
perdita dei capelli, la diarrea, un aumento della pressione del sangue o l’osteoporosi.
Se i farmaci determinano la remissione della malattia possono essere ridotti od
eventualmente sospesi.
Sono comunque in corso continue sperimentazioni cliniche che servono per sperimentare nuovi trattamenti,
dato che sino ad ora non esiste ancora un farmaco che possa guarire dal LES.
LA SINDROME DI SJOGREN
La sindrome di Sjogren (SS) è una malattia infiammatoria cronica sistemica, a patogenesi autoimmunitaria, che colpisce tipicamente le ghiandole
esocrine dell’organismo. Viene classificata fra le connettiviti.
La secchezza oculare (xeroftalmia) e la secchezza orale (xerostomia) ne costituiscono i sintomi più
comuni.
La malattia ha una prevalenza di 5-10 casi/100.000 abitanti/anno.
Vi è una netta predilezione per il sesso femminile con un rapporto femmine/maschi pari a 9:1.
Come viene classificata?
La malattia può essere distinta in:
PRIMARIA: caratterizzata dal coinvolgimento delle ghiandole esocrine (“sindrome sicca”) con o senza impegno sistemico;
SECONDARIA: quando è associata ad altre malattie autoimmunitarie (quali per esempio artrite reumatoide, LES, pclerodermia, vasculiti, connettivite mista, cirrosi biliare
primitiva, tiroidite di Hashimoto, crioglobulinemia mista, Polimiosite, colite ulcerosa, morbo di Chron ed altre).
Da cosa è causata e come si sviluppa?
Come nelle altre malattie autoimmunitarie, l’eziologia della SS è verosimilmente di tipo multifattoriale per il concorso di fattori genetici,
ormonali, immunologici e virali, anche se non tutti identificati con precisione.
L’importanza dei fattori genetici è confermata dall’associazione tra SS ed alcuni fenotipi dell’HLA:
- DRW52
- DR3: forma associata ad anticorpi anti-SSA e -SSB
- DR4: forma associata ad artrite reumatoide
La netta prevalenza del sesso femminile può essere verosimilmente attribuita all’influenza degli estrogeni, che aumentano l’attivazione policlonale dei
linfociti B e la formazione di autoanticorpi (attivano il sistema immune).
Vi sono molti studi in merito alla correlazione fra SS ed infezioni; in particolare il Citomegalovirus (CMV) ed il
virus di Ebstein Barr (EBV) sono considerati possibili induttori della malattia. Questi virus hanno infatti un facile accesso (tropismo) alle ghiandole salivari e potrebbero innescare le
reazioni autoimmuni verso le stesse, sia con un meccanismo di attivazione aspecifica policlonale B linfocitaria sia con un meccanismo di mimetismo molecolare, cioè inducendo una risposta
autoimmunitaria verso antigeni virali capace però di coinvolgere anche strutture self (appartenenti cioè all’organismo).
Per quanto riguarda la patogenesi la SS è caratterizzata da un espansione policlonale dei linfociti B (una
attivazione dei linfociti di tipo B) e da un’ipergammaglobulinemia con presenza di autoanticorpi. Gli autoanticorpi mediano il danno alle ghiandole lacrimali provocando la
distruzione del dotto secretorio mentre a livello delle salivari provocano un’ingrossamento dei dotti escretori con successiva atrofia e distruzione della ghiandola stessa. Alterazioni
analoghe possono verificarsi a livello di tutte le ghiandole dell’organismo con conseguente secchezza della cute, della vulva, dell’albero bronchiale, della gola e della mucosa
nasale.
Come si manifesta?
L e manifestazioni cliniche all’esordio possono essere aspecifiche e comparire molti anni prima della diagnosi definitiva.
Manifestazioni principali sono i segni e sintomi di secchezza orale ed oculare. Spesso sono presenti poi sintomi costituzionali quali astenia, affaticabilità, dolori muscolari ed articolari diffusi, febbricola, linfoadenopatie.
Le manifestazioni oculari si manifestano con bruciore oculare, prurito, arrossamento, sensazione di corpo estraneo o di “sabbia negli occhi” e fotofobia;
altri sintomi sono la difficoltà a leggere ed a guardare la televisione; obiettivamente si rileva un quadro di cheratocongiuntivite secca, non specifico per la Sindrome di Sjögren.Le
complicanze oculari comprendono: la congiuntivite infettiva (stafilococcica), le ulcerazioni della cornea, l'uveite posteriore.
L’interessamento orale è legato ad una riduzione della secrezione salivare ed è responsabile della sensazione
di secchezza orale, dell’ alterazione del gusto, dell'aumento di carie dentarie, della difficoltà nella masticazione soprattutto dei cibi secchi con necessità di assumere liquidi.
Nel 60% dei casi si rileva obiettivamente un ingrossamento delle parotidi, che all'esordio di malattia, può essere monolaterale e in seguito divenire bilaterale.
Benché nella maggior parte dei casi l’interessamento rimane limitato alle ghiandole salivari e/o lacrimali,
in alcuni pazienti vi può essere l’interessamento delle mucose nasali (crostosità, epistassi, alterazione dell’olfatto); del tratto respiratorio superiore ed inferiore (secchezza
tracheale, bronchiti); dell’apparato genitale esterno femminile (prurito e secchezza vaginale) e dell’apparato gastrointestinale (gastrite cronica atrofica).
ALTRE MANIFESTAZIONI
- OSTEOARTICOLARI: nelle forme con impegno sistemico o generalizzato sono frequenti le artralgie o le artriti (generalmente poliarticolari,
simmetriche, simili all’artrite reumatoide; se ne differenziano per la buona riposta agli antinfiammatori e per la non erosività).
- CUTANEE: il fenomeno di Raynaud si presenta nel 35% dei pazienti, accompagnato da un aspetto edematoso delle mani, in assenza di ulcere digitali. Possono
manifestarsi anche porpora ipergammaglobulinemica ed eritema anulare (che è una lesione eritematosa rilevata, con area centrale pallida, localizzata al volto, agli arti superiori ed al
dorso), oltre a secchezza cutanea, spesso associata a prurito ed a ridotta sudorazione.
- ENDOCRINE: particolarmente frequente è l’impegno tiroideo con gozzo eutiroideo o tiroidite autoimmune (che porta generalmente ad ipotiroidismo).
- NEUROLOGICHE: neuropatia periferica agli arti inferiori (si manifesta generalmente con intorpidimento e sensazione di formicolio e bruciore agli arti). Temibile è
l’interessamento del sistema nervoso centrale con vasculiti ad interessamento dei vasi cerebrali che clinicamente si manifestano con sintomi e segni legati all’area cerebrale
interessata.
- POLMONARI: oltre alla tosse secca, vi possono essere complicanze serie (sebbene più rare), quali l’ipertensione polmonare o l’interstiziopatia polmonare.
- RENALI: l’interessamento è poco frequente. La nefropatia tubulo-interstiziale è caratterizzata da diffusa infiltrazione linfocitaria dell’interstizio renale
capace di coinvolgere ed alterare la funzione tubulare; tale lesione si esprime clinicamente con l’acidosi tubulare renale.
- DISORDINI LINFOPROLIFERATIVI: il rischio di linfoma a cellule B nei pazienti affetti da sindrome di Sjogren è 44 volte superiore rispetto alla popolazione
generale. Possono essere linfomi, sia a carico delle ghiandole salivari che extraghiandolari, a cellule B indifferenziate o ben differenziate e linfomi a cellule T.
Come sono i parametri di laboratorio?
Reperti spesso presenti sono una anemia normocromica normocitica secondaria all’infiammazione cronica ed un
aumento degli indici di flogosi. Nel 20% dei pazienti vi può essere leucopenia, cioè una riduzione della conta dei globuli bianchi. Fattore reumatoide IgM ad alto titolo, immunocomplessi
circolanti e crioglobuline sono presenti nella maggior parte dei pazienti. I marker sierologici della SS sono gli anticorpi anti-Ro/SSA e gli anti-La/SSB. Gli anticorpi antinucleo sono
presenti in circa l’80% dei casi.
Quali sono le indagini diagnostiche?
Le secrezioni lacrimali si valutano con prove funzionali semplici:
TEST DI SCHIRMER: valuta la secrezione lacrimale tramite una striscia di carta assorbente messa a contatto con la ghiandola lacrimale inferiore. Il test risulta positivo
se, dopo 5 minuti, la striscia è imbibita meno di 5 mm.
BREAK-UP TIME TEST (BUT) o tempo di rottura del film lacrimale: si esegue colorando il film corneale con fluoresceina e osservando la sua rottura con lampada a fessura.
Un tempo di rottura inferiore a 10 secondi è da considerarsi patologico.
TEST AL ROSA BENGALA: il rosa bengala è un colorante che può essere impiegato per identificare i danni della superficie oculare.
Le procedure diagnostiche usate per la valutazione dell’impegno salivare le seguenti:
BIOPSIA DELLE GHIANDOLE SALIVARI MINORI: è altamente specifica per Sindrome di Sjögren se il prelievo comprende 5-10 ghiandole, con il tessuto connettivo
circostante; il tessuto presenta tipicamente infiltrati linfocitari.
SCIALOGRAFIA: si esegue introducendo un mezzo di contrasto idrosolubile nel dotto di Stenone. Si osservano tipicamente dilatazione, restringimenti del dotto di Stenone o
dei dotti principali, oppure microcalcificazioni e marcata ritenzione del mezzo di contrasto dopo stimolo acido (succo di limone).
ECOGRAFIA PAROTIDEA: evidenzia le alterazioni con buona sensibilità ma scarsa specificità.
RISONANZA MAGNETICA NUCLEARE: molto specifica.
Come si può porre la diagnosi?
I criteri classificativi utilizzati sono i seguenti:
I. Sintomi oculari: una risposta positiva ad almeno una delle seguenti domande:
- Ha una sensazione giornaliera e fastidiosa di secchezza oculare da almeno 3 mesi?
- Ha una sensazione ricorrente di sabbia negli occhi?
- Fa uso di lacrime artificiali più di tre volte al giorno?
II. Sintomi orali: una risposta positiva ad almeno una delle seguenti domande:
- Ha una sensazione giornaliera di secchezza orale da almeno 3 mesi?
- Ha avuto in età adulta episodi ricorrenti e persistenti di ingrossamento delle ghiandole salivari?
- E' costretto a bere frequentemente quando mangia cibi secchi?
III. Segni oculari: evidenza di impegno oculare documentato dalla positività di almeno uno dei seguenti test:
- Test di Schirmer
- Test al Rosa Bengala
IV. Istopatologia: rilievo di scialoadenite focale linfocitaria, valutata da un istopatologo esperto, a livello delle ghiandole salivari minori definito da
un focus score >1 (Focus: aggregato di almeno 50 cellule mononucleate in prossimità di un acino apparentemente intatto; Focus score: numero di foci per 4 mm2)
V. Impegno delle ghiandole salivari: evidenza di impegno delle ghiandole salivari documentato dalla
positività di almeno uno dei seguenti test:
- Scintigrafia salivare (riduzione della captazione, della concentrazione e lenta eliminazione del tracciante)
- Scialografia parotidea (restringimenti e dilatazioni diffuse a livello dei dotti principali)
- Flusso salivare non stimolato( <1.5 ml in 15 min)
VI. Autoanticorpi: presenza nel siero dei seguenti autoanticorpi:
- anti-Ro (SSA) o -La(SSB)
REGOLE PER LA CORRETTA CLASSIFICAZIONE:
La presenza di 4 o più criteri in pazienti senza malattie potenzialmente associate alla S. di Sjögren è
indicativo di SINDROME DI SJÖGREN PRIMARIA.
Nei pazienti con malattie potenzialmente associate alla Sindrome di Sjögren la positività del punto I o II
più quella di almeno 2 fra i punti III-VI è probante per una SINDROME DI SJÖGREN SECONDARIA
Quale la diagnosi differenziale?
La diagnosi differenziale va posta con:
- altre cause di xerostomia (farmaci: anti-ipertensivi, antidepressivi ed ipno-inducenti) e di xeroftalmia (ostruzione dei dotti lacrimali, utilizzo di lenti a contatto)
- forme di Sindrome di Sjögren secondarie
- linfoma
- crioglobulinemia di tipo II, infezione da HCV
- AIDS
Quali sono i principi terapeutici?
Terapia preventiva
- Igiene orale, visite oculistiche periodiche, umidificazione degli ambienti.
Terapia sintomatica
- Per il trattamento della secchezza oculare è consigliato l’uso di LACRIME ARTIFICIALI a base di metilcellulosa o di polivinil alcol.
- Per il trattamento della secchezza orale è consigliata la frequente assunzione di liquidi, l’uso di spray o gel idratanti del cavo orale.
- In casi selezionati può essere usata anche la pilocarpina cloridrato (Salagen) che aumenta la secrezione ghiandolare.
Terapia sistemica: viene utilizzata nelle forme con interessamento multi-organo. Si basa sull’utilizzo di:
- anti-infiammatori non steroidei
- steroidi a bassa dose
- idrossiclorochina (nelle forme più lievi di malattia con impegno articolare, febbre, rash cutaneo)
- immunosoppressori (methotrexato ed azatioprina vengono riservati ai casi di grave impegno d’organo)
- Rituximab: farmaco biologico riservato a casi selezionati(vedi sezione farmaci biologici)
In sintesi la sindrome di Sjogren è una connettivite che causa in particolare un impegno infiammatorio delle ghiandole esocrine dell'organismo. I disturbi principali sono legati alla secchezza dell’occhio e della bocca anche se può esserci secchezza tracheale, vaginale ed ad altri distretti. E’ legata ad una attivazione autoimmune. E’ una malattia sistemica e può coinvolgere anche altri distretti dell’organismo oltre le ghiandole esocrone. Va segnalato inoltre il maggior rischio di sviluppare un linfoma B (tumore delle ghiandole linfatiche). Per questo motivo e per il possibile coinvolgimento sistemico il paziente va seguito attentamente. La terapia può essere sintomatica o in relazione ai vari tipo di impegno d’organo.
SCLERODERMIA
La sclerosi sistemica è una malattia infiammatoria sistemica del tessuto connettivo caratterizzata da una vasculopatia diffusa e da accumulo di collageno (fibrosi) e di altri componenti della matrice connettivale a carico della cute e di vari organi interni: apparato gastroenterico, cuore, polmone, rene, articolazioni e muscoli. Sclero-dermia significa letteralmente pelle-dura per rendere visivo l’aspetto del paziente.
Epidemiologia
La reale diffusione della SSc non è ben definita. La malattia colpisce i soggetti di tutte le età, ma esordisce più frequentemente fra i 20 ed i 40 anni. Sono colpite maggiormente le donne, con un rapporto F/M oscillante nei vari studi da un minimo di 3/1 ad un massimo di 15/1. Sono stati segnalati dati di incidenza oscillanti fra 0.85 e 19 nuovi casi/milione di individui/anno, e di prevalenza fra 20 e 250 casi/milione di individui.
Classificazione
La SSc presenta una notevole variabilità sul piano dell’espressività clinica, della gravità e dell’evolutività. Sono stati, quindi, operati vari tentativi tesi a classificare la malattia in sottogruppi differenti sul piano clinico-prognostico. Per lunghi anni è stata utilizzata una classificazione in 2 sottotipi: sindrome CREST (da calcinosi, Raynaud, esofagopatia, sclerodattilia, teleangectasie) e SSc con sclerodermia diffusa. Una classificazione riassuntiva è espressa nella tabella seguente:
Tabella 1: classificazione della sclerodermia
|
Sclerosi sistemica |
cutanea diffusa (dcSSc) cutanea limitata (lcSSc) cutanea intermedia (icSSc) |
|
sclerodermia localizzata |
lineare, morfea |
|
sclerodermia associata ad altre connettiviti |
sindrome overlap |
|
sclerodermia indotta da sostanza tossiche |
cloruro di vinile, solventi organici, silicone, farmaci (bleomicina, pentazocina) |

La sclerosi sistemica si distingue in base all’estensione dell’interessamento cutaneo. Nella forma diffusa il decorso è spesso rapido con coinvolgimento di tutta la superficie corporea ed elevata frequenza di impegno viscerale. La forma limitata corrisponde alla già definita sindrome CREST. Nella forma intermedia la sclerosi cutanea colpisce anche la parte prossimale degli arti ma risparmia il tronco. Esiste inoltre una SSc “sine sclerodermia” e una forma “in fase presclerotica” in cui il quadro cutaneo non è evidente classicamente ma possono essere presenti gli altri rilievi clinici tipici della malattia.
Eziologia
L’eziologia è ignota, la patogenesi è a tutt’oggi non ben precisata. Alcune evidenze suggeriscono l’implicazione di fattori predisponenti individuali. Uno di questi è rappresentato dal sesso femminile. Sono state, inoltre, segnalate alcune associazioni significative con antigeni del sistema maggiore di istocompatibilità (predisposizione genetica): il DR3, il DR5, DR1, A1, B8. Sono stati, infine, descritti alcuni casi di SSc in componenti della stessa famiglia. La fibrosi, che costituisce la caratteristica più evidente della malattia, è da considerare un elemento a valle della cascata patogenetica. Essa è dovuta ad aumentata produzione di collageno (I, III, IV) e di altri costituenti della matrice (fibronectina, glicosaminoglicani) da parte dei fibroblasti. Le piastrine e le mastcellule sembrano anch’esse svolgere un ruolo nella stimolazione dei fibroblasti. Le prime producono molti fattori di crescita, soprattutto, il PDGF (Platelet Derived Growth Factor), che ha una notevolissima attività di stimolazione dei fibroblasti e delle cellule muscolari lisce. Le mastcellule, che sono presenti in numero elevato nella cute sclerodermica in fase precoce di malattia, producono istamina che stimola la sintesi di collageno e la proliferazione dei fibroblasti ed ha azione endotelio-lesiva. Particolare attenzione è rivolta al momento dagli studiosi del problema al Transforming Growth Factor beta (TGF-beta). Questo fattore di crescita, prodotto come il PDGF da monociti e piastrine, stimola fortemente la sintesi di collageno dei fibroblasti e aumenta l’espressione di RNA messaggero per il collageno in essi. La vasculopatia sclerodermica presenta due elementi caratteristici: la microangiopatia obliterativa e la alterazione proliferativa delle piccole arterie. Sin dalle fasi iniziali è possibile osservare un danno endoteliale diffuso, la cui genesi non è nota. Il danno endoteliale, comunque causato, determina alti livelli sierici di fattore VIII-fattore di von Willenbrand antigene e promuove l’attivazione delle piastrine, testimoniata dalla presenza di aggreggati piastrinici circolanti e di alti livelli di beta-tromboglobulina plasmatica con probabile liberazione di fattori di crescita (TGF-beta, PDGF). Il terzo elemento della verosimile triade patogenetica della SSc è costituito dalle alterazioni della risposta immune. Il reperto, già in fase iniziale prefibrotica di infiltrati linfocitari perivascolari, costituiti essenzialmente da T linfociti, a livello della cute e degli altri organi colpiti e la dimostrazione di ipersensibilità al collageno IV pongono in discussione una patogenesi immunologica da immunità cellulo-mediata nei confronti del collageno o di altri costituenti. Nella SSc sono inoltre presenti molti reperti indicativi di una risposta immune umorale alterata: ipergammaglobulinemia con iperIgG, fattori reumatoidi, crioglobuline, immunocomplessi circolanti, anticorpi anti-cardiolipina, anticorpi antinucleari.

Fisiopatologia
Negli ultimi anni sono stati pubblicati numerosi studi a sostegno dell’implicazione di processi ischemici transitori nella genesi del danno tissutale nella SSc. Alcuni Autori, con una terminologia altamente evocatrice del processo, hanno parlato di fenomeno di Raynaud a carico degli organi interni. In questa luce va vista la segnalazione strettamente clinica di una maggiore incidenza di crisi renale sclerodermica nei mesi invernali, così come l’aumento dell’attività reninica plasmatica e la riduzione della perfusione renale indotti dall’esposizione al freddo. Studi analoghi a carico del polmone hanno permesso di evidenziare che il freddo induce un aumento della pressione arteriosa polmonare ed una riduzione della perfusione. Con l’utilizzazione della scintigrafia cardiaca al tallio sono stati dimostrati difetti di perfusione sia fissi che reversibili. Il freddo induce anche alterazioni transitorie della mobilità delle pareti cardiache e della contrattilità del ventricolo sinistro.
Quadro clinico
Nella grande maggioranza dei casi la prima manifestazione della SSc è costituita dal fenomeno di Raynaud. Negli altri pazienti la malattia esordisce per lo più con manifestazioni articolari, edema alle estremità, sclerosi cutanea. In alcuni casi occorrono contemporaneamente due manifestazioni d’esordio (Raynaud più edema o Raynaud più artrite o Raynaud più sclerosi cutanea). L’esordio con il solo fenomeno di Raynaud costituisce la regola nella lcSSc (forma localizzata), mentre nelle forme diffuse non è raro che altre manifestazioni rappresentano il primo segno della connettivite. L’intervallo di tempo intercorrente fra la comparsa del fenomeno di Raynaud e quella della sclerosi cutanea è estremamente variabile. Esso è breve, intorno ad un anno o meno, nella dcSSc (diffusa), mentre è piuttosto lungo, anche decadi, nella lcSSc.
Per fenomeno di Raynaud si intende un evento vasospastico parossistico, scatenato dal freddo o da stati emozionali e caratterizzato, nella sua espressione classica trifasica, da pallore (fase ischemica), cui segue cianosi (fase asfittica) e rossore (fase iperemica reattiva). Coesistono sul piano subiettivo torpore e parestesie. Le mani rappresentano la sede più frequentemente colpita ma possono essere interessati i piedi, i padiglioni auricolari, il naso, la lingua. Un fenomeno di Raynaud è presente nella quasi totalità dei pazienti di SSc, ma può occorrere in molte altre condizioni o decorrere in maniera isolata (Raynaud primario o malattia di Raynaud). Il Raynaud sclerodermico è più frequentemente bi- o trifasico e condiziona spesso l’insorgenza di alterazioni trofiche ai polpastrelli sotto forma di necrosi, escare, ulcere e cicatrici residue. Alcuni Autori hanno riferito che nei casi di SSc senza Raynaud sono pìù frequenti la nefropatia e la miocardiopatia. Questi casi, pertanto, hanno una prognosi peggiore.
L’aumento di consistenza della cute (sclerodermia) costituisce l’aspetto clinicamente più evidente della malattia. In una piccola percentuale di pazienti esso non è presente, realizzandosi un quadro di “SSc sine sclerodermia”. Sono classicamente descritte tre fasi evolutive delle alterazioni cutanee sclerodermiche: edematosa, indurativa o sclerotica, atrofica. Il primo stadio non si manifesta in tutti i pazienti. Quando presente, l’edema interessa le dita, le mani e/o i piedi, e può estendersi a parti prossimali degli arti e del volto. La cute è aumentata di spessore e di consistenza, tesa e lucida. I solchi sono spianati. Alla digitopressione non si forma, se non raramente, una fovea (infossamento della cute). Nella fase sclerotica il reperto è tipico. La cute è aumentata di consistenza, tesa, aderente ai piani sottostanti, a tratti lucida. I peli sono diradati o assenti. La sclerosi è variamente estesa nei pazienti dei diversi sottogruppi di SSc raggiungendo la massima estensione nello spazio di 2-3 anni dalla sua comparsa. L’aumento di consistenza della cute raggiunge gradi diversi nei vari pazienti ed in ciascun paziente nelle varie parti del corpo. Sono stati elaborati alcuni sistemi per valutare in modo quantitativo l’entità della sclerosi cutanea. Il più utilizzato è quello proposto dal gruppo di Rodnan. Questi autori valutano l’aumento di consistenza della cute su una scala di 4 livelli (0 = assente; 1 = lieve; 2 = moderato; 3 = severo;) in 17 distretti, ottenendo così uno score totale con un valore massimo di 51. La misurazione di tale score può essere utile per monitorare l’attività della malattia e la risposta ad eventuali terapie. L’aspetto del paziente sclerodermico nella fase sclerotica è classico. Il volto è amimico, le labbra sono assottigliate (microcheilia), l’apertura della rima orale è ridotta (microstomia). In alcuni casi, dopo anni di decorso, si possono realizzare aspetti di mento corrugato e di labbra a borsa di tabacco. L’aderenza della cute ai tessuti sottostanti nelle regioni articolari e periarticolari è causa di contratture in flessione alle mani (aspetto ad “artiglio”), ai gomiti, ecc. In questi distretti la pressione esercitata dall’osso sottostante sulla cute tesa, con l’eventuale intervento di traumi, può determinare la comparsa di lesioni, che vanno incontro a sovrainfezioni di difficile trattamento. La fase atrofica tardiva si accompagna a lassità, fragilità e diminuzione di consistenza della cute ed a ricomparsa dei peli. In alcuni casi una melanodermia (colorazione brunastra) diffusa (che non interessa le mucose a differenza del morbo di Addison) accompagna o precede il realizzarsi della sclerosi cutanea. In altri si realizzano chiazze di pseudovitiligo che si alternano a zone iperpigmentate. Un reperto molto frequente è costituito dalle teleangectasie. Esse rappresentano uno dei 5 elementi della pentade CREST e sono state ritenute caratteristiche delle forme a sclerosi cutanea limitata, ma, in realtà, possono presentarsi in pazienti di tutti i sottogruppi di SSc, essendo solo più fonde e più numerose nella lcSSc. Le regioni più caratteristicamente colpite sono il volto e la parte superiore del torace ma sono interessati anche altri distretti cutanei e le mucose. A livello del vallo dell’unghia è frequente il reperto di teleangectasie cosiddette cuticolari. Le calcificazioni, intra- e sottocutanee, occorrono classicamente nella lcSSc, ma possono presentarsi anche negli altri sottogruppi di SSc, anche se sono di dimensioni tendenzialmente maggiori nella cosiddetta sindrome CREST. Solo raramente esse costituiscono un reperto clinicamente apprezzabile, essendo, per lo più la loro presenza scoperta all’esame radiologico. Presentano dimensioni estremamente variabili: caratteristici sono i piccoli depositi evidenziabili a livello dei polpastrelli ma si possono realizzare conglomerati più grossi. Possono causare ulcerazioni della cute sovrastante con fuoriuscita di materiale simile a pasta dentifricia.
La frequenza di manifestazioni articolari è alta (circa il 90%) nella SSc, almeno nelle fasi avanzate. Il quadro è quello di una poliartrite in una minoranza di casi (10%); più frequentemente si tratta di poliartralgie con o senza rigidità o di sola rigidità articolare. Il decorso è quasi sempre cronico o intermittente, ma in alcuni casi l’artropatia sclerodermica ha carattere episodico. Sul piano anatomo-radiologico sono presenti reperti di riduzione della rima articolare, osteoporosi iuxtaarticolare, anchilosi e di erosioni (fino al 40% dei casi) simili a quelle dell’artrite reumatoide. Sul piano anatomo-isto-patologico è presente una sinovite. Essa, però, a differenza di quella della artrite reumatoide non esita nella formazione di un panno evidente. La membrana sinoviale è sede di infiltrati infiammatori diffusi o sotto forma di aggregati, e presenta sulla sua superficie depositi di fibrina, responsabili degli sfregamenti udibili al movimento delle articolazioni all’esame obiettivo. Il liquido sinoviale, esaminato in pochi casi, ha mostrato, in pazienti con chiare manifestazioni di artrite, caratteristiche di liquido infiammatorio, ma con poche cellule (<2000/mm3) prevalentemente mononucleari. Indipendentemente dalle manifestazioni articolari in senso stretto, la limitazione funzionale delle articolazioni sclerodermiche è dovuta alla sclerosi della cute e dei tessuti circostanti, che costringe le articolazioni configurando una periartropatia caratteristica. I tendini presentano comunemente un aumento di consistenza, che èfacilmente apprezzabile a livello della regione volare del polso, dove, così come in altre sedi sono a volte apprezzabili dei tipici sfregamenti alla palpazione durante i movimenti. Il reperto di sfregamenti tendinei è significativamente più frequente nella dcSSc che nella lcSSc, tanto da essere ritenuto un reperto caratteristico delle forme diffuse. Alle ossa l’alterazione più caratteristica è costituita dall’acroosteolisi (perdita di osso), che, quando associata a calcinosi è ritenuta diagnostica della SSc. Altri reperti sono un’osteoporosi diffusa e, nel 10% circa dei casi, una periostite, soprattutto delle ossa lunghe delle mani e dei piedi. La quasi totalità dei pazienti di SSc presenta una lieve astenia muscolare diffusa, dovuta essenzialmente al disuso correlato alle manifestazioni generali o a quelle articolari della malattia. Esiste, poi, una miopatia primitiva, caratterizzata da astenia muscolare prossimale e da elevazione lieve degli enzimi (CPK, LDH). In alcuni casi una polimiosite definita decorre nei pazienti con SSc. Questi pazienti sono oggi classificati come casi di overlap fra due connettiviti, piuttosto che come SSc con miosite. La distinzione fra il secondo ed il terzo tipo di miosite sopraelencati è fondamentale, non essendo la miosite sclerodermica primitiva sensibile agli steroidi a differenza dell’altra.
Nell’ambito degli organi interni l’apparato gastroenterico è il più frequentemente colpito dalla SSc. Il cavo orale offre a considerare in aggiunta alla microcheilia ed alla microstomia, la diminuzione del volume della lingua (microglossia), la presenza di teleangectasie mucose ed una diffusa atrofia della mucosa, alla cui genesi può compartecipare la riduzione del flusso salivare dovuta ad una sindrome di Sjogren secondaria in senso stretto o a fibrosi delle ghiandole salivari. É anche comune l’occorrenza di gengivite e di riassorbimento degli alveoli con conseguente mobilità degli elementi dentari. La prima alterazione esofagea documentabile consiste in una riduzione del tono dello sfintere esofageo inferiore (LES), che può decorrere asintomatica o causare pirosi retrosternale e rigurgiti acidi. Si realizza poi una ipomotilità dei 2/3 inferiori dell’esofago, di per sè causa di disfagia (difficoltà a deglutire) “bassa” ai cibi solidi. Con il progredire della malattia la parte inferiore dell’esofago diviene dilatata ed atonica. In alcuni pazienti, inoltre, a causa del reflusso gastroesofageo acido, si realizza un’esofagite peptica, che evolve in stenosi esofagea, rendendo molto difficile l’alimentazione. Lo studio dell’esofagopatia sclerodermica si avvale della radiografia standard con pasto baritato, della manometria esofagea, della pHmetria delle 24 ore, della scintigrafia e della esofagoscopia. La manometria esofagea è molto più sensibile della semplice radiografia nell’evidenziare l’ipomotilità e l’ipotono dello sfintere esofageo inferiore, ma non è ben accettata da tutti i pazienti. La pH-metria delle 24 ore permette di documentare l’entità e la frequenza degli episodi di reflusso gastroesofageo acido. L’esofagoscopia, infine, documenta la presenza di esofagite peptica e, con l’ausilio di esami colturali, permette di mettere in evidenza una possibile candidiasi esofagea sovrapposta, che può essere opportunamente trattata. Lo stomaco è poco frequentemente colpito dalla SSc. L’intestino tenue può essere ipotonico e causare distensione, crampi addominali, diarrea cronica o intermittente e una sindrome da malassorbimento, la cui presenza può essere indagata mediante il test allo xilosio e la valutazione dei grassi fecali. All’instaurarsi di questo ultimo quadro compartecipa la proliferazione della flora batterica intestinale facilitata dall’ipomotilità che determina alterato assorbimento dei grassi e consumo di vitamina Bl2 con conseguenti quadri di anemia macrocitica. Può occorrere un ileo pseudo-ostruttivo. Sul piano diagnostico la radiografia con pasto baritato, o meglio il clisma del tenue, mettono in evidenza l’ipomotilità del viscere, la dilatazione delle anse. L’ipomotilità del colon può non causare alcuna manifestazione clinica o condizionare la presenza di stipsi o di fenomeni pseudo-ostruttivi. Sul piano radiologico è pressocchè patognomonico il reperto di diverticoli ad ampia base che solo raramente possono essere sede di perforazione o di stasi fecale. La motilità anorettale è anch’essa ridotta con la possibile comparsa di manifestazioni di prolasso rettale e di incontinenza fecale. Il fegato non è interessato dalla SSc. È frequente, invece, l’overlap fra la cirrosi biliare primitiva e la SSc, soprattutto la lcSSc.
In un’alta percentuale di casi di pazienti di SSc (70% circa) si realizza una pneumopatia interstiziale più marcata a livello dei campi polmonari inferiori. Sul piano istopatologico, nelle fasi iniziali, è presente una componente infiammatoria costituita sia da cellule mononucleate (linfociti e macrofagi) sia da cellule polimorfonucleate (neutrofili ed eosinofili). Il quadro è poi dominato dalla fibrosi. A causa della limitata attività fisica degli sclerodermici, la pneumopatia non dà sempre segni di sè. Nel 60% dei casi si registra una dispnea da sforzo. Nettamente inferiore, invece, è la prevalenza della dispnea a riposo. In circa il 10% dei pazienti è presente tosse non produttiva, che si associa ad espettorazione quando si realizza una sovrapposizione infettiva. L’esame obiettivo mette in evidenza la presenza di crepitii basali bilaterali da fibrosi interstiziale in circa il 40% dei casi. La radiografia del torace è relativamente poco sensibile nello svelare l’esistenza della pneumopatia sclerodermica. Essa mette in evidenza, in circa il 40% dei pazienti, una fibrosi interstiziale prevalente ai lobi inferiori con aspetti per lo più reticolari o reticolonodulari In alcuni casi si osservano aspetti a nido d’ape (honeycomb) che riflettono la presenza di lesioni cistiche polmonari. Più sensibile è la TAC ad alta risoluzione (HRTC). Lo studio della funzionalità respiratoria permette di documentare la presenza di alterazioni in circa il 70% dei pazienti di SSc. I reperti più frequenti sono quelli indicativi di una pneumopatia restrittiva: riduzione proporzionale della capacità vitale forzata e del flusso espiratorio massimo in un secondo, ridotta compliance polmonare cui si associa frequentemente una riduzione della diffusione alveolo-capillare. Quest’ultimo reperto può costituire, comunque un’anomalia isolata, deponendo per l’esistenza di un interessamento vascolare isolato in assenza di fibrosi interstiziale. La scintigrafia polmonare col gallio 67 mette in evidenza un’aumentata captazione soprattutto nei casi ad esordio recente nei quali è attiva una componente infiammatoria. Allo scopo di evidenziare un’alveolite è indubbiamente più utile il lavaggio broneoalveolare, che mostra un aumento dei neutrofili e degli eosinofili (è questo il reperto tipico della fibrosi polmonare primitiva), o un aumento dei linfociti e/o dei macrofagi. Nel 10% dei pazienti di lcSSc a 15-30 anni dall’esordio della malattia si realizza un’ipertensione polmonare severa da vasculopatia delle arterie polmonari di piccolo e medio calibro, a livello delle quali è presente un restringimento marcato del lume, dovuto ad aumento di spessore della parete da accumulo di connettivo nell’intima e da ipertrofia nella media. Sul piano clinico l’ipertensione polmonare causa una dispnea severa progressiva, cui segue un quadro di scompenso cardiaco destro. La diagnosi è confermata dai risultati dell’ecocardiogramma e del cateterismo cardiaco. L’evolutività dell’ipertensione polmonare maligna è spiccata: la sopravvivenza a partire dall’esordio delle manifestazioni da ipertensione polmonare si aggira intorno ai due anni. È opportuno precisare che la fibrosi interstiziale può condizionare di per sè una riduzione del letto vascolare polmonare da obliterazione dei capillari e delle arteriole delle zone fibrotiche. Ciò si traduce in un aumento della pressione arteriosa polmonare che, comunque, si differenzia sul piano fisiopatologico dall’ipertensione maligna. Accanto alle due condizioni sovradescritte va considerato il possibile interessamento pleurico, che assume le caratteristiche di una pleurite secca o essudativa e colpisce, secondo studi autoptici, un’alta percentuale di pazienti, ma si rende evidente clinicamente in circa il 15% dei casi.
Manifestazioni cliniche di pericardite sono evidenti nel 7% dei casi sotto forma di pericardite acuta o cronica essudativa. Nella maggior parte dei casi di pericardite essudativa si tratta di piccoli versamenti, documentabili con l’ecografia e privi di espressione clinica. Raramente l’entità del versamento è cospicua In questi casi la pericardite essudativa ha un valore prognostico sfavorevole. In particolare è stata notata una stretta correlazione temporale (<6 mesi) fra l’esordio di una pericardite essudativa e Io sviluppo di una crisi renale sclerodermica. L’interessamento del miocardio da parte della SSc consiste in una fibrosi miocardica frequentemente parcellare, raramente diffusa. Le arterie coronarie extramurali sono comunemente indenni; i rami intramiocardici presentano reperti di riduzione del lume da accumulo di connettivo nell’intima associato ad infiltrati infiammatori. Sul piano strettamente clinico la fibrosi miocardica può condizionare lo sviluppo di aritmie, disturbi della conduzione e scompenso cardiaco congestizio. L’utilizzazione di indagini strumentali permette di mettere in evidenza alterazioni miocardiche in un’alta percentualcdi casi clinicamente silenti. L’elettrocardiogramma standard è alterato in circa il 50% dei casi. L’alterazione più frequente è costituita da un allungamento del PR. Altri reperti sono: BAV di Il e III grado; BBD; BBS; EAS; blocchi bifascicolari; segni di ipertrofia ventricolare destra o sinistra; - extrasistoli atriali e ventricolari. Sono descritti quadri di necrosi miocardica in assenza di manifestazioni cliniche correlate. L’ECG-Holter documenta frequentemente la presenza di aritmie atriali e/o ventricolari, che invece emergono solo nel 10% dei casi all’ECG standard. L’ecocardiogramma permette di documentare alterazioni della contrattilità miocardica. La scintigrafia al tallio mette in evidenza un’alta frequenza di difetti di perfusione sia fissi (indicativi di fibrosi miocardica) sia reversibili, che sono, cioè, indotti dall’esposizione al freddo o dall’esercizio fisico e regrediscono con il ritorno alle condizioni basali.

La manifestazione caratteristica dell’interessamento del rene in corso di SSc è costituita dalla crisi renale sclerodermica o ipertensione maligna sclerodermica. Si tratta di un quadro ad esordio brusco comprendente ipertensione sisto-diastolica di tipo maligno, insufficienza renale ingravescente, disturbi visivi da retinopatia ipertensiva, cefalea, ictus cerebrale, edema polmonare acuto. In alcuni casi sono presenti anemia emolitica microangiopatica e piastrinopenia da coagulazione intravascolare. L’esame delle urine mette in evidenza inizialmente la presenza di proteinuria discreta, microematuria e a volte cilindruria. Con il progredire dell’insufficienza renale il paziente non curato va incontro ad oliguria ed anuria. Un reperto frequente è costituito dall’aumento notevole dell’attività reninica plasmatica, cui viene assegnato un ruolo nel determinismo patogenetico della condizione. La crisi renale sclerodermica colpisce essenzialmente pazienti di dcSSc, entro 3-4 anni dall’esordio della malattia, nei quali si è realizzata una rapida progressione dell’estensione della sclerosi cutanea e/o che non presentino fenomeno di Raynaud. Ricerche recenti suggeriscono che i corticosteroidi a dosi medio alte possono favorire l’insorgenza di crisi renale
Sindrome sicca: una xerostomia ed una xeroftalmia sono reperti frequenti in corso di SSc. Questo complesso sintomatologico è spesso sostenuto da fibrosi ghiandolare, ma nel 15% dei casi è presente un’infiltrazione linfocitaria delle ghiandole salivari minori, quale è tipica della sindrome di Sjogren in senso stretto. In alcuni di questi pazienti sono stati messi in evidenza anticorpi anti-SSA ed anti-SSB.
Un ipotiroidismo, per lo più subclinico, è stato messo in evidenza in circa il 25% dei pazienti di SSc. Sul piano istologico il quadro è dominato da una fibrosi.
Una sindrome del tunnel carpale è frequente in corso di SSc e può rappresentare la prima manifestazione della malattia.
Laboratorio.
Gli indici di infiammazione sono frequentemente alterati in corso di SSc. L’emocromo puòmettere in evidenza la presenza di anemia in alcuni casi da malattia infiammatoria cronica o sideropenica da esofagite peptica o da malassorbimento; in corso di crisi renale può esserci una anemia emolitica microangiopatica con piastrinopenia. La valutazione degli enzimi di colestasi è di ausilio nel sospettare i casi con cirrosi biliare associata. Può essere presente una ipergammaglobulinemia moderata ed il fattore reumatoide sierico. La positività ANA è presente in quasi tutti i pazienti. Hanno valore diagnostico il reperto di anticorpi anticentromero (ACA) che sono caratteristici della forma limitata e gli anticorpianti Scl-70 (ora definiti anti topoisomerasi I) che sono spesso presenti nella forma diffusa. Altro reperto specifico è il riscontro di anticorpi antiRNA-polimerasi III (ARA) Alla immunofluorescenza tipicamente mostrano un pattern antinucleolare, granulare diffuso o punteggiato.
Diagnosi
La diagnosi di SSc è agevole nei pazienti in cui è presente sclerosi cutanea. Vanno tenute in considerazione molte condizioni che possono comportare un aumento di consistenza della cute (vedi tabella) ma è raro che si pongano seri problemi diagnostici e che si debba ricorrere all’esame bioptico.
Tabella 2: condizioni caratterizzate da “sclerodermia”
|
Sclerosi sistemica |
|
Sclerodermia circoscritta |
|
Fascite eosinofila |
|
Malattia da cloruro di vinile |
|
Graft versus Host disease cronica |
|
Sindrome da olio tossico |
|
Scleredema di Buschke |
|
Scleromixedema |
|
Fibrosi a bleomicina |
|
Fibrosi da pentazocina |
|
Sclerodattilia dei diabetici |
|
Algodistrofie riflesse |
|
Malattia di Dupuytren |
|
Acrodermatite cronica atrophicans |
|
Malattia celiaca dell’adulto |
Nei confronti di queste condizioni, infatti, la SSc si differenzia per la simmetricità dell’interessamento della cute, per le manifestazioni a carico degli organi interni e per i reperti autoanticorpali. Qualche problema è posto dai casi di SSc sine sclerodermia. Questi pazienti vengono alla nostra osservazione per il fenomeno di Raynaud. In essi la diagnosi corretta può essere emessa quando viene dimostrata la coesistenza di altri reperti tipici della SSc: esofagopatia, acroosteolisi, calcinosi, fibrosi polmonare, anticorpi antinucleari patognomonici: anticorpi anticentromero, anti-Scl-70. Problemi diagnostici di una qualche difficoltà pone comunque la SSc al suo esordio. Il fenomeno di Raynaud, che rappresenta nella maggior parte dei casi la prima manifestazione della malattia, è comune a molte condizioni Pertanto un corretto approccio al paziente con fenomeno di Raynaud isolato deve essere teso all’analisi di altre patologie. L’utilità della capillaroscopia è oggi ben codificata. Sono tipici di una SSc reperto di megacapillari associato o meno a quello di aree avascolari. La presenza di queste ultime è ritenuta caratteristica della dcSSc. Questi reperti capillaroscopici possono essere, però, osservati in pazienti con dermatopolimiosite o con sindromi di overlap. Essi, pertanto, non hanno un significato patognomonico.
I criteri classificativi (tabella 3) sono tesi ad uniformare casistiche di centri diversi. Questi criteri presentano alcuni limiti se sono utilizzati erroneamente per la diagnosi. Infatti molte condizioni diverse dalla SSc possono presentarsi con sclerosi cutanea prossimale alle metacarpofalangee (criterio maggiore). In questi casi l’utilizzazione acritica dei criteri potrebbe fare etichettare come affetti da SSc pazienti che non lo sono. D’altro canto è da considerare il possibile riscontro in clinica di pazienti con fenomeno di Raynaud, senza ulcere o necrosi o cicatrici residue e con sclerodattilia, esofagopata ed anticorpi anticentromero (ACA). Questi pazienti sono oggettivamente affetti da SSc, ma non soddisfano criteri ARA.
Tabella 3 Criteri ARC classificativi SSc (1980)
|
CRITERIO MAGGIORE |
Sclerodermia prossimale alle metacarpofalangee |
|
CRITERI MINORI |
Sclerodattilia Necrosi o cicatrici dei polpastrelli Fibrosi polmonare bibasale |
|
* Considerate le dovute diagnosi di esclusione, la presenza del solo criterio maggiore oppure di almeno 2 criteri minori permette di classificare la condizione come sclerosi sistemica con una sensibilità del 97% ed una specificità del 98% |
|
Decorso.
Il decorso della SSc è variabile da caso a caso, soprattutto fra pazienti appartenenti a sottogruppi diversi di malattia. La sopravvivenza media si aggira intorno al 90% a 5 anni dalla comparsa della malattia ed intorno a 65% a 10 anni. La sopravvivenza è inferiore nei sottogruppi a maggiore estensione della sclerosi cutanea. Nella lcSSc la complicanza più temibile è l’ipertensione polmonare, che si sviluppa solo in una minoranza di casi e dopo molti anni dall’esordio della connettivite. Nella dcSSc la morte è dovuta nella quasi totalità dei casi alla miocardiopatia o alla pneumopatia o, meno frequentemente alla nefropatia, dalle complicante gastroenterologiche (atonia intestinale). Per il clinico è importante poter prevedere, anche se solo sul piano tendenziale, la probabile evoluzione della malattia. In linea generale si ritiene che il sesso maschile, la razza negra, la rapida diffusione e la maggiore estensione della sclerosi cutanea ed il fatto che questa preceda per tempo di comparsa il fenomeno di Raynaud sono fattori prognostici sfavorevoli. Sia pure indirettamente il reperto di anti-Scl-70, essendo associato alla dcSSc può avere un tale significato, così come è quello di ANA con pattern nucleolare o granulare diffuso. Al contrario la limitata estensione della sclerosi cutanea ed il reperto di ACA ad essa associato hanno un significato prognostico relativamente favorevole. In una minoranza di casi tutti affetti da dcSSc, l’evolutività della malattia è molto marcata e la morte interviene nello spazio di 1-2 anni dall’esordio. Questi casi erano stati da alcuni definiti “sclerodermia acuta diffusa”. L’elevata mortalità è anche legata alla prevalenza di neoplasie rilevate in questi pazienti in modo nettamente più frequente rispetto la popolazione generale
Terapia
Il trattamento farmacologico della SSc va distinto in due aspetti: terapia della malattia nella sua globalità, tesa ad interferire sulle tappe del processo patogenetico che ne è alla base; terapia delle singole manifestazioni. L’estrema variabilità nell’espressione clinica e nell’evolutività non consentono di dettare uno schema terapeutico che possa essere valido per tutti i pazienti di SSc. L’elaborazione del programma terapeutico emergerà, quindi, dalla analisi dello stadio evolutivo e dallo studio approfondito degli organi colpiti. Per quanto concerne la terapia di fondo non esiste a tutt’oggi un farmaco in grado di “curare” la SSc. Sono stati usati: griseofulvina, colchicina, D-penicillamina come farmaci interferenti con il metabolismo del collagene La penicillamina possiede inoltre attività immunomodulante. I farmaci vasodilatatori contrastano le alterazioni vascolari caratteristiche in particolare i fenomeni di vasospasmo. I più usati sono i calcio antagonisti (nifedipina in particolare), ACE inibitori e le prostacicline (iloprost). L’iloprost pare avere anche un azione antifibrotica e di miglioramento della vasculopatyia sclerodermica. Il suo uso, ad infusioni cicliche periodiche, è ampiamente utilizzato non solo per il trattamento delle complicanze ischemiche ma anche come terapia “di fondo” nella forma specie sistemica..L’introduzione in terapia degli ACE inibitori ha migliorato la prognosi della crisi renale sclerodermica. E’ migliorato il controllo dell’ipertensione arteriosa anche se rimane difficile impedire la progressione verso l’insufficienza renale. In molti pazienti è quindi necessario il ricorso alla dialisi. Numerosi farmaci immunosoppressori sono stati utilizzati. Effetti positivi sono stati descritti con la ciclofosfamide in particolare in caso di interessamento polmonare in fase precoce (alveolite). Più limitate le esperienze con azatioprina, methotrexate, ciclosporina.. Partendo dall’ipotesi che pone il danno endoteliale e la conseguente aggregazione piastrinica alla base del determinismo patogenetico della SSc, alcuni autori hanno utilizzato farmaci antiaggreganti piastrinici (dipiridamolo ed aspirina) Nei pazienti con artrite è indicato un trattamento con antiflogistici non steroidei e/o steroidi a basse dosi Molto difficile è, invece, il trattamento delle deformità da contratture in flessione. La fisiochinesiterapia può essere di qualche ausilio L’esofagopatia sclerodermica si avvale di alcune norme igieniche (mantenimento della stazione eretta per alcune ore dopo i pasti, utilizzazione di due o più cuscini durante il sonno, pasti moderati soprattutto di sera) tese a minimizzare il reflusso gastroesofageo. Sono usati, inoltre, la metoclopramide ed il domperidone. La riduzione del tono dello sfintere esofageo inferiore condiziona lo sviluppo di una esofagite peptica la cui insorgenza può essere combattuta con l’uso di inibitori della secrezione acida. L’interessamento dell’intestino tenue è teoricamente influenzato dalla metoclopramide, che in pratica, però, non cambia significativamente la motilità del viscere. Per le sindromi da malassorbimento sono utilizzati cicli di antibiotici tesi a ridurre la crescita di batteri, che interferiscono con l’assorbimento dei grassi e delle vitamine liposolubili. Il deficit alimentare va combattuto con la somministrazione di queste sostanze così come di proteine ed eventualmente di ferro e vitamina B12 nei casi in cui questi fattori risultino carenti (anemia sideropenica, macrocitica). Pleurite, pericardite, miosite rispondono generalmente agli steroidi a dosi medie. Nell’ambito delle terapie di fondo della SSc vanno anche considerate le tecniche di aferesi. Alcuni Autori hanno registrato dopo cicli di plasmaferesi miglioramenti delle manifestazioni cutanee, vascolari periferiche ed articolari. Nei casi più gravi si è provveduto al trapianto autologo di midollo nel tentativo di eliminare i cloni cellulari inducenti l’autoimmunità. In aggiunta alla terapia farmacologica il trattamento della SSc si avvale di misure generali: si consiglia di mantenere la temperatura ambientale intorno ai 25 gradi allo scopo di evitare il vasospasmo caratteristico del fenomeno di Raynaud. A temperature ambientali più basse il paziente deve coprire bene sia le estremità sia il capo e la parte centrale del corpo, inducendo il calore centrale una vasodilatazione periferica. Sono da prediligere attività lavorative che non condizionano traumi, soprattutto alle dita. Sono, infine, di qualche utilità la chinesiterapia tesa ad evitare l’instaurarsi di contratture in flessione delle articolazioni. Alcuni Autori consigliano l’utilizzazione di creme idratanti, ad esempio alla lanolina, allo scopo di combattere la secchezza delle cute.
In casi selezionati con impegno polmonare hanno infine trovato impiego farmaci biologici come il Rituximab(vedi sezione Farmaci biologici) o il Riociguat, farmaco attivo sulla ipertensione polmonare.
LE MIOPATIE INFIAMMATORIE
Le miositi sono malattie infiammatorie autoimmuni croniche della muscolatura striata, e talora della cute, ad eziologia sconosciuta, che
appartengono alla categoria delle connettiviti.
Il gruppo "storico" comprende, oltre alla Dermatomiosite [DM] (dal greco: infiammazione di pelle e muscolo) e alla Polimiosite [PM] (=infiammazione di molti muscoli) dell'adulto, le forme giovanili
(con insorgenza non oltre i 17 anni), la miosite associata ad altre connettiviti e la forma associata a neoplasie. Oltre a queste, negli ultimi anni sono state individuate altre forme: la Miosite da
corpi inclusi (chiamate così per i caratteristici riscontri bioptici; v. in seguito), ad insorgenza senile e con andamento subdolo, la sindrome anti sintetasica, caratterizzata dalla presenza di
specifici anticorpi e accompagnata da un quadro clinico caratteristico e la Dermatomiosite amiopatica che a fianco del caratteristico quadro cutaneo non manifesta interessamento clinico muscolare per
un periodo di tre anni dalla diagnosi; infine va ricordata la miosite necrotizzante, forma caratterizzata da un impegno clinico anche molto severo e dall’assenza di infiltrato infiammatorio ed una
elevata proporzione di fibre muscolari necrotiche alla biopsia.
Quale la loro frequenza?
Sono malattie rare, con incidenza che varia da 1 a 12 nuovi casi all'anno per milione di abitanti. Per rendere l’idea corrisponde ad 1/4 dei casi di Lupus Eritematoso Sistemico, a 1/2 della Sclerosi
Sistemica e ad 1/50 dell'Artrite Reumatoide. La prevalenza varia da 5 ai 10 casi per 100000 abitanti.
In realtà, proprio per il fatto che si tratta di malattie rare, e quindi ancora poco conosciute, è probabile che tali dati siano sottostimati, dal momento che un significativo numero di pazienti
verosimilmente sfugge alla diagnosi e alla ospedalizzazione.
Sebbene le miopatie infiammatorie possano insorgere in qualsiasi età sono stati osservati due picchi, uno sotto i 20 anni di età ed uno in pazienti tra i 55 e i 69 anni. Il rapporto femmine: maschi è
di 2.5: 1.
Quale la causa?
La causa è tuttora sconosciuta. Numerosi dati suggeriscono una componente o almeno una predisposizione genetica a cui si sovrappongono dei fattori ambientali, infettivi (Enterovirus, HIV,
Cytomegalovirus, Parvovirus B19, HCV (virus epatite C), Borrelia burgdorferi) o tossici. Tra questi ultimi non vanno dimenticati alcuni farmaci, come ad esempio gli ipocolesterolemizzanti orali
(fibrati e statine) che negli anni scorsi hanno fatto parlare di sé proprio per alcuni casi di tossicità muscolare, che il più delle volte si manifesta con dolore muscolare ed elevazione degli enzimi
muscolari, ma nei casi più severi può arrivare ad una vera miosite.
Quali le manifestazioni cliniche?
Il sintomo guida più frequente all’esordio, e il più caratteristico, è la debolezza dei muscoli prossimali degli arti, lamentata nell’ 80 % dei casi; molto frequente è anche la debolezza generale,
mentre più rari sono altri sintomi generali come febbricola, cefalea e calo ponderale.
MUSCOLO
La debolezza muscolare, in genere simmetrica e prevalentemente a carico della muscolatura prossimale degli arti, anche se, ad eccezione dei muscoli oculomotori, possono essere coinvolti tutti i
distretti muscolari, compresi quelli della respirazione, deglutizione e fonazione. La muscolatura distale degli arti è comunque raramente interessata in corso di Dermato-Polimiosite a differenza di
quanto avviene nella Miosite da corpi inclusi. L’ esordio è in genere subdolo e va aggravandosi durante un periodo di settimane, mesi o, nel caso della miosite da corpi inclusi, addirittura
anni.
Alla debolezza possono accompagnarsi dolori muscolari diffusi nei distretti coinvolti e fatica. Il paziente diventa incapace a salire le scale, ad alzarsi dalla posizione seduta o accucciata o ad
alzare le braccia per pettinarsi o farsi la barba. La dolorabilità alla palpazione del muscolo è variabile.
Una difficoltà alla deglutizione e sintomi di aspirazione ab ingestis possono riflettere un interessamento della muscolatura striata del faringe o dell’esofago superiore.
CUTE
Specifiche della Dermatomiosite, anche se presenti solo in circa il 30 % dei casi, sono le papule di Gottron. Si tratta di papule o placche rosso-violacee, leggermente rilevate, presenti al di sopra
delle prominenze ossee, soprattutto sulla superficie estensoria delle articolazioni delle mani, sui gomiti, sulle ginocchia e sui malleoli interni delle caviglie.
Il rash eliotropo, presente in non più del 25 % dei pazienti, è caratterizzato da una colorazione bruno-violacea delle palpebre superiori accompagnata talora da edema, con distribuzione simmetrica;
secondo alcuni autori può essere correlato all’andamento clinico della malattia. Simile al precedente e presente in circa il 40 % dei casi è il “segno dello scialle”, chiamato così perché colpisce la
fronte, il collo, le spalle, il tronco e il viso. Quando la stessa manifestazione è localizzata alla regione anteriore del collo ed alla parte superoanteriore del torace prende il nome di “V sign”.
In entrambi i casi le lesioni possono risultare fotosensibili.
Lesioni meno specifiche possono prodursi al cuoio capelluto con una alopecia non cicatrizzante, di solito in concomitanza o in seguito a riacutizzazioni della miosite oppure ancora con placche
rilevate che assomigliano a lesioni psoriasiche o alla dermatite seborroica.
Il fenomeno di Raynaud (= variazione del colore della cute, in genere delle dita, che diventa prima bianca, poi bluastra ed infine rossa, in seguito a vasospasmo provocato dal freddo o vibrazioni od
emozione) è riportato in generale nel 35 % dei casi.
La calcinosi è una manifestazione molto più frequente nella dermatomiosite dell’infanzia, rispetto alla forma dell’adulto, dal momento che nei bambini e adolescenti può colpire dal 30 al 70 % dei
pazienti. Consiste iella comparsa di noduli sottocutanei di color bianco-giallastro, della grandezza di alcuni millimetri di diametro, ma estremamente dolorosi, che in genere crescono sulle
prominenze ossee o nella sede di traumi ripetuti; tali noduli sono formati da depositi di calcio che possono fuoriuscire attraverso la superficie cutanea ed essere soggetti ad infezioni
sovrapposte.
In generale il decorso delle manifestazioni cutanee non è parallelo a quello della malattia muscolare in quanto può precederla anche di anni e può persistere anche dopo il controllo o la scomparsa
della miosite, rimanendo per molto tempo l’unico segno di malattia.
Nella sindrome anti sintetasica caratteristica è la mano da macchinista (v. in seguito).
MANIFESTAZIONI ARTICOLARI
L’interessamento articolare a tipo artralgie e/o poliartrite è presente in circa il 25 % dei pazienti ed è più frequente nelle forme overlap con altre connettiviti e nella sindrome antisintetasica.
Comunemente si tratta di artralgie generalizzate, accompagnate da rigidità mattutina, anche se può occorrere un’artrite simmetrica, non deformante, di solito transitoria; mani, polsi, gomiti,
ginocchia e caviglie sono le sedi più frequentemente coinvolte. Normalmente l’impegno articolare si manifesta nelle fasi precoci della malattia e risponde comunque bene alla terapia per la
sottostante malattia muscolare.
In corso di sindrome anti sintetasica l’artrite può divenire cronica e deformante anche se raramente erosiva.
MANIFESTAZIONI POLMONARI
L’impegno polmonare varia per tipo e per gravità fino ad essere, in alcuni casi, la più importante manifestazione della malattia, che ne condiziona seriamente la prognosi. Il quadro più tipico è
quello dell’interstiziopatia polmonare (=ispessimento del tessuto polmonare tra un alveolo e l’altro tale da poter rendere difficili gli scambi gassosi), spesso associato ad un coinvolgimento
esofageo ed alla presenza di anticorpi anti sintetasi (v. in seguito)
Indipendentemente dal tipo di esordio, l’interstiziopatia polmonare può precedere, seguire od insorgere contemporaneamente all’impegno muscolare ed in generale la presenza e la severità del quadro
polmonare non è correlata alla gravità del quadro muscolare.
ALTRE MANIFESTAZIONI
La disfagia (difficoltà alla deglutizione) da interessamento della muscolatura della deglutizione e/o del terzo prossimale dell’esofago (a muscolatura striata) è presente in circa 1/3 dei casi e può
complicarsi con aspirazione nell’albero tracheo-bronchiale e comparsa di polmoniti ab ingestis. Tale coinvolgimento è correlato con la severità dell’impegno muscolare ed è responsivo alla terapia
steroidea.
Manifestazioni cardiache possono presentarsi in oltre il 50 % dei casi ma solo una piccola percentuale di pazienti è sintomatica. Anormalità elettrocardiografiche sono evidenti nel 40 % dei
pazienti.
I disturbi del ritmo e della conduzione sono i più frequenti seguiti dalla cardiomiopatia.
ALTRE MANIFESTAZIONI
L’associazione tra neoplasia e miosite è sempre stata motivo di controversie fin dalla pubblicazione del primo caso nel 1916. La presenza di neoplasia è segnalata nel 7-30 % dei pazienti e
l’associazione è certa con la DM, ma non con la PM. Il rischio di neoplasia inoltre è maggiore in pazienti con un’età maggiore di 50 anni al momento della diagnosi di DM. La natura paraneoplastica
troverebbe conferma nelle segnalazioni di risoluzione delle manifestazioni cutanee e muscolari in seguito a trattamento della neoplasia, e dalla loro ricomparsa in caso di recidiva. Tutti i tipi di
neoplasia possono risultare correlati, riflettendo la prevalenza nella popolazione della stessa età, ma sembra esistere una maggior frequenza di neoplasie ovariche.
I fattori che devono indurre il sospetto di neoplasia sono l’età avanzata, il sesso femminile, la presenza di una malattia resistente alla terapia e recidivante, in particolare se con rash cutaneo
esteso e atipico o con prurito severo. Molti autori raccomandano l’esecuzione di una mammografia ed una valutazione pelvica, completa di visita ginecologica, ecografia transvaginale e controllo
routinario del CA 125 nelle donne, uno screening per il cancro della prostata nell’uomo più una radiografia del torace e una valutazione dell’addome in tutti i pazienti.
Miosite da corpi inclusi
Considerata una polimiosite a tutti gli effetti fino agli anni ’70, in seguito è stata tenuta distinta da questa, venendo riconosciuta come entità specifica per le sue peculiarità cliniche e
istopatologiche. L’esordio è particolarmente insidioso con una latenza media di circa 6 anni tra la comparsa dei sintomi e la sua diagnosi. Colpisce prevalentemente pazienti al di sopra dei 50 anni
con un rapporto maschi - femmine di 2:1.
Il sintomo principale è la debolezza, che coinvolge sia la muscolatura prossimale che distale degli arti, con un precoce interessamento dei flessori dell’avambraccio, della mano e del quadricipite.
Si arriva a difficoltà della deambulazione con frequenti cadute. L’atrofia muscolare è un riflesso della cronicità della malattia e costituisce spesso un aspetto tipico di questi pazienti. Un’altra
manifestazione frequente è la difficoltà alla deglutizione che colpisce oltre il 60 % dei soggetti durante il decorso della malattia.
Il livello del CPK sierico varia da valori normali ad un aumento fino a 5 volte il valore massimo della norma, mentre l’elettromiografia dimostra un quadro tipico di miosite.
La diagnosi comunque viene confermata dalla biopsia.
Sindrome antisintetasica
E’ caratterizzata dalla presenza di anticorpi anti sintetasi, associata ad un esordio acuto di Polimiosite/Dermatomiosite, interstiziopatia polmonare e, nella maggior parte dei casi, febbre, “mani da
meccanico”.poliartrite non erosiva e fenomeno di Raynaud
Tra gli anticorpi l’anti Jo-1 è in assoluto il primo, il più noto e il più comune anticorpo associato a questa malattia, dal momento che viene riscontrato in circa il 20-30 % dei pazienti con PM e
nel 2-10 % di quelli con DM. Inoltre è l’unico che viene testato routinariamente. Oltre a questo si conoscono altri anticorpi anti sintetasi che, tutti insieme, si riscontrano in non più del 10 % dei
pazienti con miosite; i più noti sono: l’anti PL-7, l’anti PL-12, anti OJ, anti EJ e l’anti KS. Sembra che la sindrome anti sintetasica classica, con tutte le manifestazioni, non sia associata
indistintamente a tutti gli anticorpi anti sintetasi: alcuni infatti, come l’anti EJ, sembrano più frequentemente associati con le manifestazioni cutanee della DM, mentre l’anti PL-7, l’anti PL-12 e
l’ anti KS sembrano più associati alla presenza di isolata fibrosi polmonare o artrite, in assenza di altre manifestazioni di miosite.
L’interstiziopatia polmonare, presente nel 50-100% dei pazienti, è la manifestazione che governa la prognosi in quanto è associata con una mortalità del 40 %. La progressione verso la fibrosi è la
regola in assenza di trattamento specifico. Le sue caratteristiche comunque non differiscono da quelle della interstiziopatia polmonare in corso di PM.
La mano da meccanico, presente in circa il 71 % dei pazienti con sindrome anti sintetasica, è una caratteristica manifestazione delle dita delle mani, che si presentano con cute ipercheratosica e
fissurazioni localizzate prevalentemente sulla superficie laterale. Non è specifica di questa sindrome in quanto è stata riscontrata, anche se con minor frequenza, in pazienti sia senza che con altri
anticorpi.
Come viene posta la diagnosi?
La diagnosi si basa sulla presenza di manifestazioni cliniche tipiche associate a riscontri laboratoristici, strumentali e bioptici.
TEST DI LABORATORIO
I test di laboratorio utilizzati per la diagnosi ed il monitoraggio delle miositi possono essere divisi in 3 categorie:
1. test per il controllo dello stato generale del paziente e delle sue condizioni cliniche (VES, PCR, emocromo,
ecc.)
2. misurazione della concentrazione degli enzimi e di sostanze derivate dal muscolo (CPK, miosina). Il CPK in particolare è aumentato in circa l’80-90 % dei pazienti con malattia attiva, con
valori di circa 10 volte il valore massimo normale, anche se, nei casi più severi, può arrivare fino a 100 volte il valore normale. La valutazione dei livelli di CPK è un metodo tuttora molto in uso
per valutare l’attività di malattia; inoltre il più delle volte anticipa di qualche settimana la sintomatologia clinica: i livelli di CPK infatti si riducono da 3 a 8 settimane prima del
miglioramento della forza muscolare e si elevano 5-6 settimane prima di una riattivazione della malattia.
3. test immunologici: gli ANA sono positivi nel 50-80 % dei pazienti e il pattern nucleare “punteggiato” è il più frequente. Alcuni ENA,come gli anti-PM-Scl, gli anti RNP e gli anti Ro/SSA si
trovano soprattutto nelle forme di miosite in overlap con altre connettiviti, in particolare con la Sclerosi sistemica e con la sindrome di Siogren. Oltre a questi, che si ritrovano comunemente anche
nelle altre connettiviti, vi sono anticorpi specifici per la miosite: anticorpi anti sintetasi, di cui si è già parlato a proposito della sindrome ad essi associata (v.sopra). Anti SRP, presenti nel
4-5 % dei pazienti soprattutto in corso di PM sono correlati a malattia severa, con esordio acuto, e frequente coinvolgimento cardiaco, scarsa risposta alla terapia e frequenti recidive con la
diminuzione della posologia, ciò che determina una prognosi estremamente severa. Infatti la sopravvivenza a 5 anni, riportata in alcune casistiche, è intorno al 25 %. Gli anti Mi2, sono segnalati nel
4-5 % e si associano tipicamente alla DM sia giovanile – nella quale costituiscono l’anticorpo miosite specifico di più comune riscontro – che dell’adulto. Le manifestazioni cutanee risultano spesso
il quadro dominante e l’aspetto più difficile da trattare. Ciò nonostante i pazienti con questi anticorpi di solito rispondono bene alla terapia ed hanno una prognosi favorevole.
ELETTROMIOGRAFIA
E’ una tecnica molto sensibile anche se non molto specifica per miosite. Quando tipica, mostra una caratteristica triade di alterazioni.
BIOPSIA MUSCOLARE
Può dimostrarsi molto utile ai fini diagnostici ed è l’unica indagine che permette di porre diagnosi di miosite da corpi inclusi; tuttavia il carattere spesso focale (cioè a macchia di leopardo)
dell’interessamento muscolare può causare la comparsa di falsi negativi.
Il quadro istopatologico muscolare della DM è differente da quello della PM, sottendendo un differente meccanismo patogenetico: nella prima l’infiltrato infiammatorio è costituito prevalentemente da
linfociti B, anziché linfociti T, e si localizza soprattutto in sede pervasale (=attorno ai vasi) e nei setti inter-fascicolari (=tra i fascicoli muscolari), piuttosto che all’interno dei fascicoli
stessi; queste caratteristiche inducono a pensare che la degenerazione delle fibre muscolari, nella DM, sia secondaria al danno microvascolare (la deplezione capillare è un evento precoce), mentre
nella PM l’epicentro è primitivamente muscolare.
Nella Miosite da corpi inclusi il quadro è molto simile a quello della PM ma vi sono le tipiche inclusioni granulari eosinofile (per questo è chiamata da corpi inclusi) nei nuclei e nel citoplasma
delle cellule muscolari.
Infine nella miosite necrotizzante si riscontra un’elevata proporzione di fibre muscolari necrotiche a fronte di un infiltrato infiammatorio modesto o assente. La inclusione di questa forma nelle
miositi autoimmuni è confermata dalla espressione sulle fibre non necrotiche dell’antigene di istocompatibilità MHC di classe I oltre che dalla risposta alla terapia steroidea.
[Il sistema MHC di classe I permette il riconoscimento di cellule estranee
all’organismo (e quindi destinate ad essere eliminate) da quelle proprie: le cellule muscolari di individui sani, così come quelle di
pazienti affetti da miopatia non infiammatoria non manifestano, cioè non presentano sulla loro parete cellulare, l’MHC di
classe I. Viceversa tutte le fibre muscolari con evidente infiammazione, ma anche le cellule muscolari non ancora invase (dalle cellule
dell’infiammazione) esprimono sulla loro parete cellulare questo complesso. Infatti in queste malattie l’induzione di MHC I nelle fibre muscolari
precede l’infiltrazione cellulare.
L’espressione di MHC I è presente nel 67% delle biopsie di pazienti con DM, nel 61% di PM, nel
96% di Miosite da corpi inclusi e solo nell’ 11% di pazienti con distrofie e 4% in pazienti con disturbi neuromuscolari]
In pratica la diagnosi specifica del tipo di miosite si basa oggi specialmente sul tipo di istologia ed immunoistochimica (espressione MHC classe I) riscontrato nella biopsia muscolare. Esistono dei
precisi criteri classificativi internazionali per le singole miositi. Questi criteri peraltro peccano di specifità sovradiagnosticando in particolare la polimiosite. Esiste un gruppo di miositi
infiammatorie “non classificabili”.
RISONANZA MAGNETICA
Lo studio in RM del muscolo è molto utile per valutare l’estensione dell’interessamento muscolare, l’attività di malattia ed il danno indotto. E’ possibile evidenziare sia uno stato di edema del
muscolo, suggestivo di flogosi o di necrosi muscolare e quindi di attività di malattia, sia l’atrofia muscolare e l’infiltrazione adiposa del muscolo come misura del danno muscolare. Inoltre è da
segnalare che la RM può essere utile per individuare le zone interessate dalla malattia per poi effettuare una biopsia muscolare “mirata”.
VALUTAZIONE DELL’IMPEGNO POLMONARE
La radiografia convenzionale del torace non è in grado di evidenziare alterazioni fino agli ultimi stadi di malattia, quando ormai l’interstiziopatia ha raggiunto il grado di fibrosi polmonare;
pertanto tale esame non è di aiuto nella diagnosi, né nel monitoraggio.
L’HRCT (TC torace ad alta risoluzione) invece è una tecnica estremamente sensibile per lo studio dell’impegno polmonare. Con tale tecnica è possibile distinguere, analogamente a
quanto rilevabile nell’interstiziopatia polmonare associata ad altre connettiviti e nelle forme idiopatiche, diversi tipi di lesioni, alcune delle quali, come l’aspetto a “vetro smerigliato”, sono
ritenute espressione prevalente di alveolite in fase attiva, mentre altre, come l’irregolarità dei margini pleurici, l’ispessimento dell’interstizio e soprattutto il cosiddetto polmone “ad alveare”
sono espressione di quadri di fibrosi già costituita.
La valutazione dell’impegno polmonare comprende anche prove di funzionalità respiratoria con diffusione del CO) alle quali possono affiancarsi indagini radioisotopiche (scintigrafia polmonare con
gallio, studio della clearance alveolare, ecc) e, in casi selezionati, la valutazione, previa broncoscopia, del liquido di lavaggio bronchiolo-alveolare per tipizzare il tipo di alveolite
presente.
Quale la diagnosi differenziale?
Quale la prognosi?
Le più comuni forme di debolezza muscolare, dalle quali le miositi infiammatorie devono essere differenziate, sono quelle dovute ad alterazioni del sistema nervoso o della giunzione neuromuscolare
(ad es. le malattie autosomiche recessive che portano a degenerazione progressiva delle cellule delle corna anteriori del midollo, la sclerosi laterale amiotrofica, la miastenia gravis) e tutto il
gruppo delle malattie metaboliche del muscolo; in particolare con queste ultime la diagnosi differenziale può essere più difficile, tanto che talvolta nemmeno la biopsia muscolare è dirimente.
Comunque verso queste patologie orientano la storia familiare e clinica, l’esordio lento ed insidioso e l’assenza di segni sistemici.
Un altro gruppo importante in diagnosi differenziale è quello delle malattie endocronologiche ed in particolare la forma legata ad ipotiroidismo.
La prognosi è diversa a seconda del tipo di forma. A grandi linee si può dire che:
1. Le forme overlap (forme associate ad altre connettiviti) hanno buona risposta terapeutica e buona
prognosi
2. La DM ha buona risposta alla terapia e moderata prognosi
3. La PM ha discreta risposta alla terapia e moderata prognosi tendendo maggiormente a recidivare
4. La miosite associata a neoplasia ha moderata risposta alla terapia; la prognosi è legata alla neoplasia sottostante
5. La miosite da corpi inclusi ha scarsa risposta alla terapia e scarsa prognosi
Quale la terapia?
L’iniziale approccio terapeutico è un punto critico determinante sia per il successivo decorso che per la prognosi. I cortisonici rimangono il trattamento di scelta. La dose iniziale utilizzata varia
in funzione della severità della malattia e del rischio di tossicità per il paziente. Il regime terapeutico più consolidato prevede la somministrazione orale giornaliera di dosi elevate (0.5-1
mg/kg/die di Prednisone) per almeno 1-3 mesi, fino ad ottenere una normalizzazione del CK (di solito in 1-2 mesi) ed il miglioramento clinico. Solo nei casi più lievi si può iniziare con dosi
inferiori a 0.5 mg/kg/die. La posologia viene in seguito ridotta del 20-25 % ogni 3-4 settimane fino ad arrivare ad una dose di mantenimento di 5-10 mg/die. In caso di riacutizzazione non si deve
tornare alla dose iniziale, ma è consigliabile aumentare fino al dosaggio minimo che permette il controllo della malattia. Nei casi severi o con gravi manifestazioni extramuscolari, come l’impegno
polmonare o cardiaco, in cui è necessario un più rapido controllo della malattia, vengono utilizzati steroidi e.v. ad alte dosi.
Per la frequente mancata risposta anche a dosi elevate, o per la comparsa di inaccettabili effetti collaterali della terapia steroidea, o per l’impossibilità di ridurre la dose di steroide a causa di
riaccensioni della malattia, viene di solito aggiunto un farmaco immunosoppressore, spesso già al momento della diagnosi.
Il methotrexate (10-15 mg/kg/settimana per os o i.m.) o l’azatioprina (1.5-2 mg/kg/die), da soli o tra loro in combinazione, sono le opzioni terapeutiche più utilizzate, mentre la ciclosporina (3-5
mg/kg/die) gode di buona considerazione non solo per il trattamento della miosite, ma anche della interstiziopatia polmonare.
La ciclofosfamide (1-2 mg/kg/die) viene, in genere, riservata al trattamento dell’interessamento polmonare. Le immunoglobuline e.v. ad alte dosi (IVIg) possono risultare efficaci soprattutto nei casi
di DM, nelle forme giovanili e in pazienti con immunodeficienza, anche se l’efficacia sembra essere di durata relativamente breve e tende a ridursi nel tempo. Pertanto l’utilizzo viene raccomandato
solo in situazioni acute, in attesa che altre terapie facciano effetto.
In casi molto severi con particolare impegno respiratorio e cardiaco può essere usata la plasmaferesi.
Un altro farmaco che ha dato buoni risultati in casi refrattari è il micofenolato mofetil; l’uso dei farmaci biologici e di Immunoglobuline umane ad alto dosaggio è riservato a casi
selezionati.
Per le manifestazioni cutanee il farmaco più usato è l’idrossiclorochina, spesso associata a steroidi per via sistemica o topica.
Un discorso a parte va fatto per la miosite da corpi inclusi, malattia con una risposta talmente scarsa alla terapia, che alcuni autori consigliano addirittura di evitare qualsiasi trattamento. Più
prudentemente però la maggior parte di essi consiglia di effettuare un tentativo con prednisone (0.5 mg/kg/die) associato a methotrexate (10 mg/settimana), almeno nei casi in cui la condizione non
sia troppo avanzata ed il rischio degli effetti collaterali non troppo elevato. Altri autori raccomandano di somministrare anche sostanze anabolizzanti e coenzima Q (30 mg per 3 volte/die) e di
associare un programma di esercizi isometrici per mantenere il più possibile la performance muscolare.
E’ utile la terapia fisica?
E’ ancora tema di dibattito se l’esercizio fisico possa peggiorare o meno il danno in un muscolo già sede di infiammazione; comunque, anche se non tutti gli autori sono concordi, il riposo è
raccomandato durante i periodi di infiammazione attiva. Restano indicati gli esercizi passivi per mantenere l’escursione articolare ed evitare contratture, in particolare nei giovani. Durante la fase
di remissione, può essere iniziata la chinesiterapia attiva.
RIASSUNTO
MIOSITI = malattie infiammatorie croniche autoimmuni della muscolatura striata, e talora della cute.
Malattie rare: incidenza da 1 a 12 nuovi casi all’anno per milione di abitanti
prevalenza da 5 ai 10 casi per 100000
abitanti.
CAUSA: sconosciuta; probabile predisposizione genetica a cui si sovrappongono fattori ambientali (infettivi, tossici, ecc)
CLINICA: debolezza muscolare, simmetrica e prevalentemente a carico della muscolatura prossimale degli arti (anche se potenzialmente tutti i distretti muscolari possono essere
impegnati); esordio subdolo e lento. Possibili dolori muscolari.
Cute: papule diGottron (placche rosso-violacee, leggermente rilevate, sopra alle prominenze ossee); rash eliotropo (colorazione bruno-violacea delle palpebre superiori con possibile edema); se il
rash è a fronte, spalle, collo e volto si chiama segno dello scialle; se al decolteè si chiama V sign; Fenomeno di Raynaud; calcinosi; mano da macchinista.
Altro: interstiziopatia polmonare (impegno molto importante che spesso condiziona la prognosi); artrite o artalgie; disfagia; disturbi del ritmo cardiaco.
Miosite da corpi inclusi: esordio sopra i 50 anni; esordio lento, interessamento anche della muscolatura distale degli arti; quadro bioptico specifico; scarsa risposta alla terapia
Sindrome anti sintetasica: presenza di anticorpi anti sintetasi, esordio acuto, interstiziopatia polmonare, febbre, mani da meccanico, poliartrite non erosiva, fenomeno di Raynaud.
Associazione a neoplasia: nel 7-30 % dei pazienti con miosite, in particolare in quelli con dermatomiosite e con età elevata; spesso l’esordio della neoplasia è successivo alla miosite, anche di
molti mesi.
DIAGNOSI: basata su manifestazioni tipiche associate a riscontri laboratoristici (CPK, anticorpi), strumentali (Elettromiografia, Risonanza Magnetica) e bioptici (fondamentale per la
diagnosi e anche per caratterizzare il sottotipo). Importante è indagare l’impegno d’organo (in particolare l’interessamento polmonare che spesso condiziona la prognosi).
DIAGNOSI DIFFERENZIALE: con tutte le forme di debolezza muscolare, in particolare malattie del sistema nervoso o della giunzione neuromuscolare, malattie metaboliche, malattie
endocrine (soprattutto ipotiroidismo)
PROGNOSI: varia a seconda delle diverse forme e dell’impegno polmonare
TERAPIA: steroidi ad alte dosi restano il trattamento di prima scelta; altri farmaci utili sono: Methotrexate, Azatioprina, Ciclosporina, Ciclofosfamide, Immunoglobuline e.v.,
Plasmaferesi, Micofenolato mofetil.
Studio medico:
Casa di Cura Villa del Rosario
Via Flaminia 499 00191 Roma
per appuntamenti tel. 06-33010257
email: pittoni2011@gmail.com
Libera Professione Intramuraria: Ospedale Sant'Andrea Via di Grottarossa 1035-1039 00189 Roma
per appuntamenti: 06-33775651/06-33776113(lu-ve ore 14-18). Cup Regionale: 06-9939 tasto 2
(sezione libera professione lu-ve ore 07.30-19.30; sa ore 7.30-13)